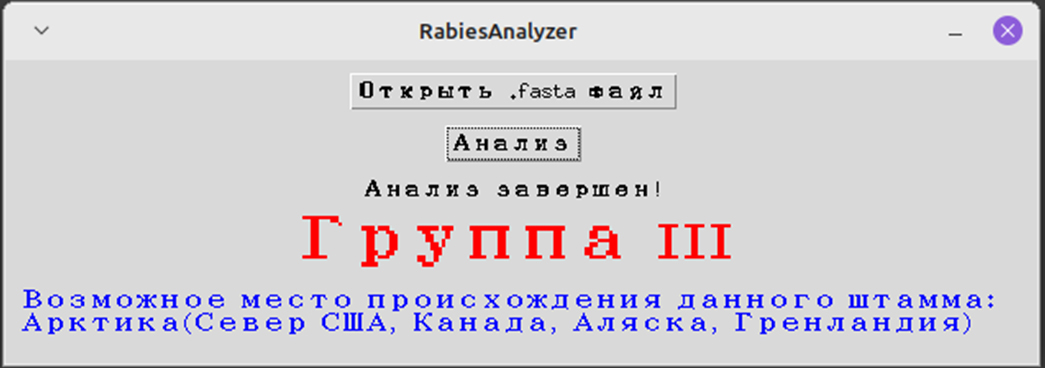
The program developed in the Russian Federation, KlebsiellaAnalyzer allows typing and analysis of whole-genome sequencing data of clinical isolates of Klebsiella pneumoniae. The programming languages Java and Python were used for software development.
The program’s ability to determine the type of capsular antigen based on batch data from whole-genome sequencing. The analysis program includes:
- Genes rmpA, rmpA2 are responsible for hypermucoid properties. The detection of one or both genes indicates that the genome under study belongs to the hypermucoid genotype. In the absence of the rmpA and rmpA2 genes, the strain under study should be classified as the classical type.
- Determination of the presence of virulence genes: 14 virulence genes are included in the program: magA, allS, mrkD, cf29a, fimF, uge, kfuA, kfuB, kfuС, kvgA, ureA2, copA2, nikA2, yciC.
- Genes for yersiniobactin synthesis: ybtS, ybtA, irp1, irp2, ybtT, psn.
- Aerobactin synthesis genes: iucA, iucC, iutA, iutA2, entA, entB, entE, fepG, fepE, fepA, pfeA_2, fes_3. Salmohelin synthesis gene iroB.
INDEL markers: the program includes 19 proprietary INDEL markers for comparative typing of the studied strains.
To detect the presence of extrachromosomal elements, the program includes genes encoding replicons of plasmids found in various world and Russian strains of Klebsiella pneumonae: IncHI1B (230kb), repB (230kb), incFII (95kb), IncFIB (230kb), IncK (54-224kb), ColRNAI, Col440I, repE_IncFIA(HI1)(95kb), IncR. During the analysis, the number of detected replicons corresponds to the number of plasmids in the strain under study.
The program provides CRISPR typing: CRISPR are special loci consisting of direct repeating sequences separated by spacers. The program includes two types of CRISPR identified in Klebsiella pneumoniae strains. The presence of the type and size of the CRISPR cassette in the genomes of the studied strains is assessed by software.